

Ecocardiograma transtorácico (ECOTT) e Ressonância Magnética Cardíaca (RMC) com espessura (Qualquer critério positivo):
● ≥ 15 mm no VE na ausência de causa secundária que justifique o achado.
● ≥ 13-14 mm no VE E familiar com CMH ou teste genético positivo.

PRINCIPAIS GENES RELACIONADOS
| MYH7 | ACTC1 |
| MYBPC3 | TNNT2 |
| TNNI3 | CSRP3 |
| TPM1 | TNNC1 |
| MYL2 | JPH2 |
| MYL3 |

Faça aqui o download do cartaz em PDF!
Todos os critérios são obrigatórios:
● Dilatação das câmaras cardíacas: Ecocardiograma transtorácico (ECOTT) e
Ressonância Magnética Cardíaca (RMC) com DDVE ≥ 58mm em homens e
≥ 52mm em mulheres.
● Disfunção ventricular: FEVE < 45% OU fração de encurtamento < 25%.
● Exclusão de outras causas: prova isquêmica negativa (se >40 anos),
sorologia chagas negativa, ausência de valvopatias importantes (exceto
insuficiência mitral secundária) E ausência de outras causas prováveis
(etilismo).
PRINCIPAIS GENES RELACIONADOS
| BAG3 | SCN5A | JPH2 |
| DES | TNNC1 | NEXN |
| FLNC | TNNT2 | TNNI3 |
| LMNA | TTN | TPM1 |
| MYH7 | DSP | VCL |
| PLN | ACTC1 | |
| RBM20 | ACTN2 |

Faça aqui o download do cartaz em PDF!

Todos os critérios são obrigatórios:
● Dilatação das câmaras cardíacas: Ecocardiograma transtorácico (ECOTT) e
Ressonância Magnética Cardíaca (RMC) com DDVE ≥ 58mm em homens e
≥ 52mm em mulheres.
● Disfunção ventricular: FEVE < 45% OU fração de encurtamento < 25%.
● Exclusão de outras causas: prova isquêmica negativa (se >40 anos),
sorologia chagas negativa, ausência de valvopatias importantes (exceto
insuficiência mitral secundária) E ausência de outras causas prováveis
(etilismo).
PRINCIPAIS GENES RELACIONADOS
| PKP2 | PLN | |
| DSG2 | FLNC | |
| DSC2 | DES | |
| DSP | LMNA | |
| JUP | CDH2 | |
| TMEM43 | RYR2 |

Faça aqui o download do cartaz em PDF!
● Ressonância magnética cardíaca (RMC) com proporção miocárdio não-compactado / compactado > 2,3 na diástole.
● Exclusão: atletas, gravidez, achado incidental de não-compactação sem contexto clínico.
PRINCIPAIS GENES RELACIONADOS
| MYH7 | LDB3 |
| MYBPC3 | TBX5 |
| TTN | NKX2-5 |
| ACTC1 | HCN4 |
| RYR2 | TAZ |
| PRDM16 |

Faça aqui o download do cartaz em PDF!

● Enchimento ventricular prejudicado com
padrão restritivo pelo Ecocardiograma
transtorácico (ECOTT) e Ressonância
Magnética Cardíaca (RMC). Ausência de
dilatação ventricular – ECOTT ou RMC.
● Ausência de hipertrofia – ECOTT ou RMC.
PRINCIPAIS GENES RELACIONADOS
| MYH7 | TTR |
| TTN | FLNC |
| ACTC1 | GLA |
| TNNI3 | TNNT2 |

Faça aqui o download do cartaz em PDF!

PRINCIPAIS CC E ETIOLOGIA GENÉTICA
Aneuploidias:
Trissomia do 13 (Sd Patau)
Trissomia do 18 (Sd Edwards)
Trissomia do 21 (Sd Down)
Monossomia do X (Sd Turner)
CNVs:
Microdeleção 22q11.2 (Sd DiGeorge)
Microdeleção 7q11.23 (Sd Williams)
Sd Cat eye
Deleção 1p36
Monogênicas:
Sd Adams-Oliver (ARHGAP31, DLL4 ,
NOTCH1, RBPJ, DOCK6, EOGT)
Defeito do Septo Atrial (GATA4, NKX2.5,
TBX5)
Defeito do Septo Atrioventricular (CRELD1,
NR2F2)
Estenose Aórtica Supravalvar (ELN)
Estenose Aórtica Valvar (NOTCH1, TAB2)
Tetralogia de Fallot (NOTCH1, FLT4,
TFAP2B)
Rasopatias (Sd Noonan, Cardio-facio-
cutâneo, NF1)
Sd CHARGE (CHD7, SEMA3E?)
Sd Alagille (JAC1, NOTCH2)
Sd Holt-Oram (TBX5)
Sd Kabuki (KMT2D, KDM6A)
Sd Ivemark (GDF1)
Heterotaxia (ACVR2B, CFC1, NODAL,
CCDC11, CFAP53, PKD1L1, ZIC3)
Sd de Smith-Lemni-Opitz (DHCR7)

Faça aqui o download do cartaz em PDF!
● Diagnóstico clínico de fibrilação atrial isolada: presença de FA em paciente sem cardiopatia estrutural, sem comorbidades e idade < 60 anos.
● Exclusão: hipertireoidismo, uso de drogas (álcool, tabagismo, drogas ilícitas).
PRINCIPAIS GENES RELACIONADOS
| SCN5A | TTN |
| KCNQ1 | KCNA5 |
| KCNH2 | GJC1 |
| TBX5 | NPPA |
| GJA5 | LMNA |
| MYL4 |

Faça aqui o download do cartaz em PDF!
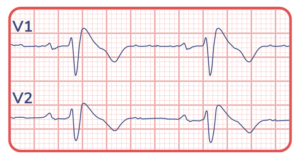
● Supra ST com morfologia de SBr1 de amplitude ≥ 2 mm em pelo menos 1 derivação direita, V1 ou V2 em posição padrão ou em derivações superiores (colocadas no 2o , 3o ou 4o espaços intercostais), sendo espontâneo ou induzido.
● Exclusão: causas secundárias de alteração de ST: isquemia miocárdica, distúrbios eletrolíticos, intoxicações por drogas
ou uso de medicação com efeito bloqueador de canal de sódio.
PRINCIPAIS GENES RELACIONADOS
| SCN5A | SCN3B |
| CACNA1C | SCN10A |
| CACNB2 | KCNE3 |
| CACNA2D1 | KCNJ8 |
| SCN1B | KCND3 |
| SCNB2 | KCNE3 |

Faça aqui o download do cartaz em PDF!
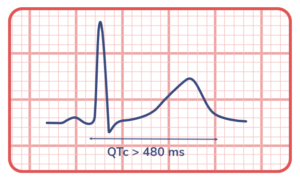
● Score de Schwartz ≥ 3,5.
● Presença de mutação patogênica em genes associados à SQTL. QT corrigido (QTc) ≥ 480 ms em ECGs repetidos.
● QTc 460-479 associado a síncope inexplicada.
○ Excluir causas secundárias de prolongamento do QT como hipocalemia, hipomagnesemia, uso de medicações que
prolongam a repolarização, como alguns antibióticos, antieméticos, etc.
Para maiores informações consulte:
www.crediblemeds.org
PRINCIPAIS GENES RELACIONADOS
| KCNQ1 | KCNJ5 |
| KCNH2 | CACNA1C |
| SCN5A | AKAP9 |
| CALM1 | TRDN |
| CALM2 | CAV3 |
| CALM3 | ANK2 |
| KCNJ2 | SCN4B |
| KCNE1 |

Faça aqui o download do cartaz em PDF!
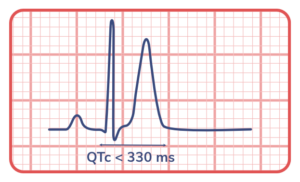
● QT corrigido (QTc) < 330–340 ms.
OU
● QTc entre 340-360 ms associados a um abaixo:
○ variante patogênica em gene associado a SQTC, história familiar de SQTC, Morte Súbita Cardíaca (MSC) abaixo de 40 anos ou sobrevida após um episódio de TV/FV na ausência de doença cardíaca.
○ QTC Score ≥ 4
○ Excluir causas secundárias de encurtamento do QT como
hipercalemia, hipercalcemia, isquemia aguda, uso de drogas ou medicações que possam encurtar a repolarização.
PRINCIPAIS GENES RELACIONADOS
| KCNH2 | KCNJ2 |
| KCNQ1 | SLC4A3 |

Faça aqui o download do cartaz em PDF!
● Disfunção do sistema de condução elétrica com
início de apresentação clínica em idade relativamente jovem e sem causa evidente.
● Excluir: cardiopatia estrutural, doença de Chagas, degeneração fibrótica, isquemia, doenças infiltrativas, valvopatias, tumores ou disfunção tireoidiana. Excluir uso de medicações cronotrópicas negativas, como beta bloqueadores, bloqueadores de canais de cálcio e antiarrítmicos.
O ECG pode demonstrar bradicardia sinusal, pausas sinusais, bloqueio sinoatrial, bloqueios atrioventriculares de variados graus e bloqueios de ramo até bloqueio
atrioventricular total.
PRINCIPAIS GENES RELACIONADOS
| LMNA | PRKAG2 |
| DES | TNNI3K |
| DMD | NKX2-5 |
| DMPK | TBX5 |
| EMD | MYL4 |
| LAMP2 | SCN5A |
| GLA | TRPM4 |
| ZNF9 |

Faça aqui o download do cartaz em PDF!
PRINCIPAIS GENES RELACIONADOS
| KCNQ1 | SCN5A |
| KCNH2 | RYR2 |
Faça aqui o download do cartaz em PDF!
● Probando < 40 anos:
○ coração estruturalmente normal + ECG normal + TV bidirecional ou
○ EEVV polimórficas ou
○ TV polimórfica induzida por exercício ou catecolaminas
● Presença de mutação patogênica
PRINCIPAIS GENES RELACIONADOS
| RYR2 | TRDN |
| CASQ2 | TECRL |
| CALM1-3 | KCNJ2 |

Faça aqui o download do cartaz em PDF!

● Variante patogênica em heterozigose no gene COL3A1.
● Se análise molecular não for conclusiva, anormalidades na síntese e mobilidade das cadeias de colágeno tipo III em fibroblastos.
PRINCIPAIS GENES RELACIONADOS
| COL3A1 |

Faça aqui o download do cartaz em PDF!

● Variante patogênica em heterozigose em um dos genes associados à SLD, E
● 1. Dilatação de raiz aórtica (z ≥2.0) ou
● 2. Dissecção tipo A ou
● 3. Achados sistêmicos:
○ Vasculares: tortuosidades arteriais (principalmente cranio-
cervicais), aneurismas e dissecções.
○ Dismorfismos craniofaciais: hipertelorismo ocular, estrabismo, úvula bífida, fenda palatina, craniossinostose.
○ Esqueléticos: pectus excavatum ou carinatum, escoliose,
hipermobilidade articular, aracnodactilia, pé torto, alterações cervicais.
○ Dermatológicas: pele aveludada, translucente, fragilidade capilar,
cicatrizes distróficas.
○ Alergo-Imunológicas: alergias alimentares, alergias sazonais,
asma, eczema, esofagite eosinofílica, doença inflamatória intestinal.
PRINCIPAIS GENES RELACIONADOS
| TGFBR2 | TGFB2 |
| TGFBR1 | SMAD3 |
| TGFB3 | SMAD2 |

Faça aqui o download do cartaz em PDF!
Na ausência de história familiar:
● Dilatação da raiz da aorta (Z-score ≥2) E ectopia lentis.
● Dilatação da raiz da aorta (Z-score ≥2) E Escore Sistêmico ≥ 7 pontos Escore Sistêmico de Marfan
● Presença de variante patogênica em heterozigose no gene FBN1 já descrita em SM, associada a ectopia lentis ou dilatação da aorta.
Obs.: as manifestações da SM podem surgir na vida adulta, logo, indivíduos com menos de 20 anos que não preenchem critérios clínicos devem ser reavaliados posteriormente.
PRINCIPAIS GENES RELACIONADOS
| FBN1 |

Faça aqui o download do cartaz em PDF!

● AAT: aumento de 50% de diâmetro da aorta em relação ao esperado em exame de imagem.
● DAT: presença de flap separando falsa luz da luz verdadeira em exame de imagem.
PRINCIPAIS GENES RELACIONADOS
| TGFB2 | TGFBR2 |
| MYH11 | TGFBR1 |
| SMAD3 | PRKG1 |
| FBN1 | MYLK |
| ACTA2 | LOX |

Faça aqui o download do cartaz em PDF!
● No Brasil o critério diagnóstico mais utilizado é o Dutch MEDPED, que segue uma escala de pontuação:
○ > 8 pontos: Definitivo
○ > 6 a 8 pontos: Provável
○ > 3 a 5 pontos: Possível
PRINCIPAIS GENES RELACIONADOS
| LDLR | PCSK9 |
| APOB | LDLRAP1 |

Faça aqui o download do cartaz em PDF!
● Xantomas: depósitos de gordura que podem estar localizados em regiões como calcanhares, joelhos, cotovelos, mãos, entre outros.
● Xantelasmas: depósito de gordura na região das pálpebras.
● Alterações hematológicas: plaquetopenia, anemia e presença de estomatócitos.
● DAC precoce (homens < 55 anos e mulheres < 60 anos). Resposta adequada ao tratamento com ezetimibe.
● Níveis de beta-sitosterol ≥ 15μg/mL.
PRINCIPAIS GENES RELACIONADOS
| ABCG5 | ABCG8 |

Faça aqui o download do cartaz em PDF!
● Dismorfismos típicos: hipertelorismo ocular, ptose palpebral, fendas palpebrais oblíquas para baixo, pescoço curto e/ou alado, alteração de pectus (em S, carinatum ou
excavatum), hipertelorismo mamilar, criptorquidia.
● Baixa estatura
● Cardiopatia congênita, mais comumente estenose valvar pulmonar, comunicação interatrial (CIA) e/ou miocardiopatia hipertrófica.
● Atraso do desenvolvimento de grau variável
● Displasia linfática dos pulmões, intestinos e/ou membros inferiores
● Problemas de coagulação
● História familiar sugestiva
● Para NF1: manchas café-com-leite, efélides axilares /inguinais, neurofibromas, tumores.
● Para síndrome de Legius: manchas café-com-leite, efélides axilares / inguinais
PRINCIPAIS GENES RELACIONADOS
| BRAF | SOS1 |
| KRAS | SOS2 |
| MAP2K1 | LZTR1 |
| MRAS | MAPK1 |
| NRAS | HRAS |
| PTPN11 | SHOC2 |
| RAF1 | PPP1CB |
| RASA2 | NF1 |
| RIT1 | SPRED1 |
| RRAS2 |

Faça aqui o download do cartaz em PDF!